
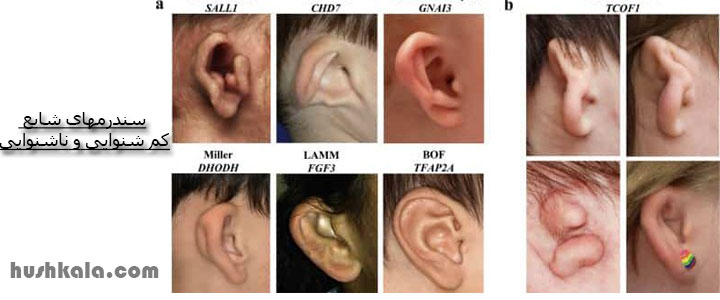
کم شنوایی سندرمیک به این معناست، که اختلال شنوایی با شرایط دیگر نیز همراه است. حدود 30 درصد از اختلالات شنوایی ارثی سندرمی هستند. به طور کلی بیش از 400 سندرم شناخته شده وجود دارد که کم شنوایی یکی از آنهاست. سندرم کم شنوایی میتواند بر سیستم های مختلف دیگر بدن از جمله کلیه ها، چشم ها و قلب نیز تاثیر بگذارد.
کم شنوایی سندرومی در یکی از الگوهای زیر به ارث می رسد:
- اتوزومال مغلوب
- اتوزومال غالب
- X مرتبط است
با ما همراه باشید تا هر کدام از این الگوها را جداگانه بررسی کنیم.
فهرست عناوین این نوشته
- 1- معرفی کمشنواییهای سندرومی اتوزومال غالب
- سندرم واردنبرگ
- سندرم برانکیو-اکولو-فیشال (Branchio-oculo-facial syndrome)
- سندرم استیکلر
- سندرم شارژه
- سندرم تریچر کالینز
- نوروفیبروماتوز تیپ 2
- 2- کمشنواییهای سندرمیک اتوزومال مغلوب
- سندروم آشر
- سندروم پندرد
- سندرم ژرول- لانژنلسون
- 3- سندرمهای مرتبط با کروموزم x
- سندرم آلپورت
- جمع بندی
1- معرفی کمشنواییهای سندرومی اتوزومال غالب
در اینجا می خواهیم شایع ترین سندرم های کم شنوایی با الگوی وراثت اتوزومال غالب را معرفی و توضیح مختصری با تاکید بر کم شنوایی آنها داشته باشیم.
سندرم واردنبرگ
- شایعترین نوع کم شنوایی در دسته اتوزومال غالب سندرمیک
- کم شنوایی یک گوش ( یکطرفه ) و یا دو گوش ( دوطرفه )
- کم شنوایی حسی عصبی
- بر روی رنگدانههای پوست، مو و چشم اثر میگذارد. افرادی که به چنین شرایطی مبتلا هستند ممکن است تکههای سفیدی بر روی پوست یا چشم خود داشته باشند که یکسان نیستند.
- پوست و چشم رنگپریده است. نشانهی دیگر آن بروز خطی از موی سفید در نزدیکی پیشانی است. در بیشتر موارد فرد ممکن است چشمانی با دورنگ متفاوت داشته باشد.
- نشانههای آن ممکن است متفاوت باشد.
- برخی از نشانهها در زمان تولد هویدا نیست و بهمرور ممکن است دیده شود.
سندرم برانکیو-اکولو-فیشال (Branchio-oculo-facial syndrome)
- اختلالی است که رشد قبل از تولد، بهخصوص رشد ساختارهایی در صورت و گردن را تحت تأثیر قرار میدهد. علائم بارز آن شامل اختلالات پوستی بر روی گردن، شکل غیرطبیعی (malformation) چشمها و گوشها و ویژگیهای متمایز در صورت است.
- کم شنوایی انتقالی ، حسی عصبی و یا میکس
- ساختار گوش خارجی ، میانی و داخلی معمولاً تغییر میکند
سندرم استیکلر
- کم شنوایی انتقالی ، حسی عصبی و یا میکس
- کم شنوایی ممکن است پیشرونده باشد ( بهمرور شدت کم شنوایی بیشتر شود)
- چشمهای برجسته، بینی کوچک و چانه عقبرفته. این کودکان معمولاً هنگام تولد کام شکافدار دارند.
سندرم شارژه
- کم شنوایی این سندروم ممکن است میکس ، حسی عصبی و یا انتقالی باشد.
- علائم مانند وجود حفره در چشم ، نقص قلبی ، عقبماندگی رشدی و تکامل ، ناهنجاری ژنیتالی و ادراری
- ناهنجاری در گوش و ساختار گوش
- شدت کم شنوایی از خفیف تا عمیق ممکن است متفاوت باشد
سندرم تریچر کالینز
- کم شنوایی از نوع انتقالی است
- علائمی ظاهری مانند فک پایین کوچک ، افتادگی بخش جانبی پلکهای پایین ، گوشهای بدشکل و یا عدم وجود لاله گوش
- اشکالات در ناحیه اطراف گوش و گونهها و شکاف کام نیز از نشانههای دیگر سندرم است.
- این سندروم از نوع اتوزومال غالب است.
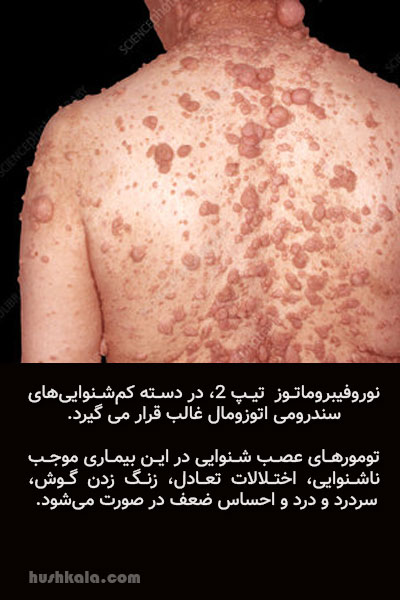
نوروفیبروماتوز تیپ 2
- یک بیماری ژنتیکی است که اعصاب و پوست را گرفتار میکند. در این بیماری تومورهای خوشخیم غیر سرطانی در مسیر اعصاب رشد میکنند و این رشد موجب بروز مشکلاتی در پوست و استخوانها میشود.
- نوروفیبروماتوز در اثر جهش در ژن تولیدکننده نوروفیبرومین ایجاد میشود. هر دو نوع بیماری ارثی و ژنتیکی بوده و ژن منتقلکننده آن غالب است
- کم شنوایی از نوع پیشرونده و حسی عصبی است و برخی از مواقع منجر به ناشنوایی کامل میشود.
- معمولاً همراه با تومورهای عصب شنوایی است و شیوع آن از هر ۲۵ هزار تولد دریکی است. تومورهای عصب شنوایی در این بیماری موجب ناشنوایی ، اختلالات تعادل، زنگ زدن گوش، سردرد و درد و احساس ضعف در صورت میشود.
- این بیماری در سنین نوجوانی یا انتهای دهه دوم عمر بروز میکند.
درصورت بروز مشکلات شنوایی می توانید به مراکز شنوایی سنجی مراجعه کنید. برای یافتن نزدیک ترین مراکز شنوایی سنجی به محل زندگی خود بر روی لیست زیر کلیک کنید.
مراکز شنوایی سنجی | آدرس و شماره تلفن کلینیک های شنوایی سنجی کشور
2- کمشنواییهای سندرمیک اتوزومال مغلوب
در اینجا نیز لیستی از سندرم های کم شنوایی با الگوی وراثت اتوزومال مغلوب را می بینید.
سندروم آشر
سندروم آشر شایعترین نوع کم شنوایی سندرمیک اتوزومال مغلوب است. سندروم آشر معمولاً باعث کم شنوایی و مشکلات بینایی فرد مبتلا میشود. اگرچه برخی از مبتلایان ممکن مشکلات بینایی و شنوایی را باهم تجربه نکنند. که دارای سه نمونه زیر است:
نوع 1
- اتوزومال مغلوب است. ( والدین هر دو باید ناقل این ژن باشند.)
- کم شنوایی عمیق، حسی عصبی
- مشکلات شدید در تعادل
- سمعک خیلی مؤثر نیست.
نوع 2
- اتوزومال مغلوب است. ( والدین هر دو باید ناقل این ژن باشند.)
- کم شنوایی متوسط تا شدید در بدو تولد
- کم شنوایی حسی عصبی
- عدم وجود مشکلات تعادلی
نوع 3
- در زمان تولد شنوایی طبیعی است
- بهمرور شنوایی بدتر میشود. حسی عصبی و پیشرونده
- مشکلات تعادلی نیز دیده میشود.
سندروم پندرد
- دومین سندروم شایع سندرمیک اتوزومال مغلوب است که منجر به کم شنوایی میشود.
- در اکثر بیماران ، کم شنوایی حسی عصبی و از نوع پیشرونده است.
- ناهنجاری در ساختار گوش داخلی ممکن است دیده شود.
- اغلب مشکلات تعادلی نیز دیده میشود.
- در برخي از موارد ، گواتر بزرگ میگردد
سندرم ژرول- لانژنلسون
- سومین سندروم شایع سندرمیک اتوزومال مغلوب است که منجر به کم شنوایی میشود.
- کم شنوایی دوطرفه، حسی عصبی و در بدو تولد دیده میشود.
- یکی دیگر از یافتههای عمده در JL-N ، ریتم قلب غیرطبیعی (طولانی Q-T) است که میتواند منجر بهاحتمال مرگ ناگهانی شود.
- ریتم قلبی غیرطبیعی میتواند از طریق درمانهای پزشکی بهخوبی درمان شود.
3- سندرمهای مرتبط با کروموزم x
در رابطه با سندرم های مرتبط با کرموزوم X می توان به سندرم آلپورت اشاره کرد.
سندرم آلپورت
- کم شنوایی از نوع پیشرونده و حسی عصبی است.
- از دیگر ویژگیهای این سندرم ، مشکلات کلیوی است
- همچنین ممکن است عدسی چشمها در افراد مبتلا دچار بدشکلی (misshapen lenses in the eyes) شده (anterior lenticonus) و رنگ غیرطبیعی در بافت حساس به نور در پشت چشم (شبکیه-retina) ایجاد شود. این ناهنجاری ها بهندرت منجر به نابینایی میشوند.
- الگوی وراثتی وابسته کروموزوم x و در برخی اتوزومال غالب و در برخی اتوزومال مغلوب است
مطالعه مقاله کاربردی زیر را به شما پیشنهاد می دهیم:
تاثیر کم شنوایی بر گفتار ، یادگیری و کودکان
جمع بندی
در این نوشته به معرفی انواع کم شنوایی های ژنتیکی سندرومی پرداختیم. بیان کردیم آنها شامل سه دسته؛ کم شنوایی ژنتیکی سندرومی با الگوی وراثت اتوزومال غالب، کم شنوایی های سندرومی با الگوی وراثت اتوزومال مغلوب، کم شنوایی های ژنتیکی سندرومی مرتبط با کرموزوم x، می باشند. همچنین شایع ترین سندروم های کم شنوایی مرتبط با هر دسته را با تاکید بر مشکل کم شنوایی آنها معرفی کردیم. امیدوارم این نوشته برای شما مفید باشد در هر صورت می توانید سوالات یا نظرات خود را در این باره در پایین در قسمت نظرات بنویسید.